Gromacs 布朗动力学模拟 Gromacs安装教程 虚拟机安装 VMware
主题简述
是研究生物大分子体系的动力学模拟软件,在国内外有着广泛的应用。目前只发布了基于Linux/Unix的安装程序包,本教程将介绍在虚拟机上安装正式版本2019.6。
工具/原料 方法/步骤
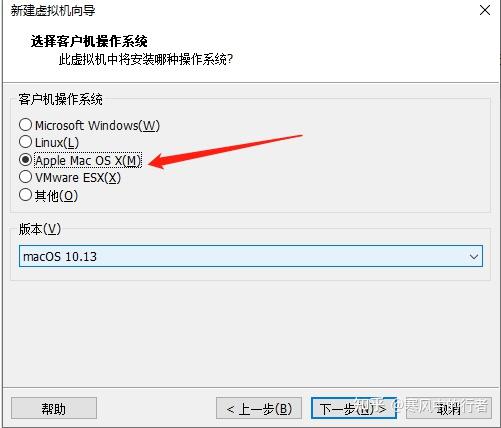
首先确保已安装好虚拟机和操作系统(如下图,无关键要的地方做了打码)。
详细安装教程
检查和安装C/C++
$ gcc -v
$ sudo yum install gcc (这里sudo是获取临时root权限,需要管理员密码;另外用"su"命令进行root操作也可以)
检查cmake:
$ cmake -version
如果没有安装cmake或版本过低,需要到网上下载,yum资源库中的cmake版本一般较低:
下载安装包:
(用系统自带的下载工具下载)
$ tar zxvf cmake-3.10.2-Linux-x86_64.tar.gz (解压安装包)$ cd cmake-3.10.2-Linux-x86_64 (进入目录)$ ./bootstrap$ make$ sudo make install
详细安装教程
安装
官网下载
下载安称耍装包:
(用辞泥系统自带的下载工具下载) (如下图)
$ tar xfz gromacs-2019.6.tar.gz (解压安装包)$ cd gromacs-2019.6 (进入目录)$ mkdir build (新建文件夹build) (如下图)$ cd build (进入目录)$ cmake .. -DGMX_BUILD_OWN_FFTW=ON -DCMAKE_INSTALL_PREFIX=/usr/local/gromacs-2019.6 (要求安装过程中自动下载FFTW库文件,指定程序安装到目录/usr/local/gromacs-2019.6)$ make (编译时间比较长)$ make check $ sudo make install (安装)
安装好后,需要设置环境变量,以便bash调用:
$ cd (回到个人目录,例如:/home/user/)$ vi .bash_profile (用vi 编辑.bash_profile文件) (如下图)在.bash_profile文档的末行添加(如下图): source /usr/local/gromacs-2019.6/bin/GMXRC 。添加完后按"Shift"+":"并输入"wq!"保存退出。重新启动bash:$ logout (注销并重新登录)
查看是否正确安装(如下图)
$ gmx - (可以看到刚才安装的版本)
查看动力学程序mdrun是否可用:
$ gmx mdrun -h
以上总结是由本人一整套从安装——安装Linux系统——系统——安装失败后的解决办法。
通过搜索“ 远程安装 Linux系统 布朗动力学模拟 分子动力学模拟分析”或者“虚拟机安装 Pro任选版本 Win Linux ”找到一家可以通过安装虚拟机解决问题的小店。
下期会提供一份Linux虚拟机安装的教程。