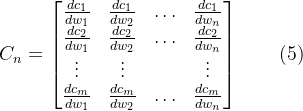药物靶标相互作用(二)
一.-DTI: A -based for drug-
基于相互作用特征的药物-靶蛋白相互作用预测神经网络 2023.5
与同样基于变压器的相比,-DTI的复杂性较低。这背后的理由是,虽然-DTI考虑了两个平行的多头注意力层,但网络的注意力头数量从8个减少到2个,设计了一个更低的隐藏层维度。因此,这大大减少了-DTI中的参数数量。这些设计有助于 -DTI 更好地拟合训练数据,同时避免由于过于复杂而导致的过度拟合。
1.模型
基于互反应特征的DTI预测双途径模型,称为-DTI。解码器经过修改,将药物和蛋白质视为两个不同的序列。
在得到蛋白质和药物的最终输入之后,解码器中添加了一个多头自注意力层来提取交互特征(下图中间的荧光色地方),从而能够学习药物和蛋白质,以及原子和氨基酸之间的复杂相互作用。解码器提取相互作用特征后,我们得到相互作用的蛋白质和药物的特征矩阵D、P,再对特征矩阵的不同维度求均值,得到给定维度中每一行的均值。最后获得的特征向量被链接并且送到分类模块进行预测。
1.药物表示
用GNN将转化为图,使用提取的化学特征的随机初始化嵌入作为GNN的初始输入。在GNN层之后,得到一个药物序列的特征向量C1,C2,C3...Cl,其中l是药物序列中的原子数。
2.蛋白质表示
蛋白质序列由20个氨基酸组成。如果我们学习一个蛋白质序列作为句子,那么组成句子的单词只有20种。为了增加特征的多样性,基于n-gram语言模型,我们将蛋白质序列中的单词定义为n-gram氨基酸。对于给定的氨基酸序列,将其拆分为重复的n-gram氨基酸序列。例如,将n设置为3,并将蛋白质序列ABCDE拆分为ABC,BCD,CDE,这样组成句子的各种单词将扩展到20的3次方。
将处理之后的序列进行D维嵌入并且初始化,然后输入门控卷积网络(包括1D卷积和GLU单元),其输出结果是蛋白质的最终表示。
2.实验 1.数据集
8:1:1切分数据集
2.参数
3.结果
测试交互模块的有效性:利用中的Davis数据集和中的标签反转数据集GPCR
复现:
AUC
human
论文 0.984 0.962 0.943
复现 0.981 0.905 0.943
论文 0.987 0.948 0.949
复现 0.986 0.930 0.958
GPCR
论文 0.82 0.699 0.796
复现 0.791 0.674 0.775
无
论文 0.810 0.704 0.768
复现 0.797 0.711 0.752
二. : A deep based multi-scale for drug-
一种基于深度学习的多尺度特征融合方法 2022.8 二区
问题:
(1).已知药物-靶点相互作用的稀缺性导致模型的应用场景有限,大多数DTI数据未标记,只有一小部分已知的DTI数据可用。此外,当我们尝试在特定病例(例如糖尿病)上实现一些DTI模型时,可用训练数据的大小甚至更小。
(2).仅限于编码分子特征,而忽略了如何对相互作用特征进行建模,提取药物和蛋白质亚结构特征,以避免上述问题,如冗余特征等。然而,最终药物-蛋白质相互作用特征的维数太大,仅从单一角度考虑,因此不够全面,无法准确预测DTIs数据的小样本。
工作:
(1)在先前的DTI预测中,仅使用其自身信息嵌入药物或蛋白质的结构,而不考虑与其他分子的全局特征。本文提出了一种在有限数据中获取更多信息的方法,以便做出准确的预测。应用谷本系数、列文施泰因距离和卷积神经网络生成药物和蛋白质全局结构相似性特征。(2)我们不仅考虑了分子的全局特征,而且还注意到药物-靶点相互作用发生在分子亚结构上。在我们的工作中,我们使用变压器来提取分子的子结构特征。(3)利用两个特征提取通道获得基于全局特征的交互作用和基于局部子结构特征的交互作用,将两个特征融合进行最终预测。
1.模型
为了得到某个药物的结构相似性特征,药物数据集里所有剩余的药物是当前药物的基准。蛋白质也如此。
由全局结构相似性特征提取通道(Sim通道)、局部结构特征提取通道(子通道)和交互预测模块(IP模块)组成。
1.Sim-利用相似性理论和卷积神经网络,对预先生成的药物/蛋白质结构相似性矩阵进行操作,获得药物/蛋白质结构相似性特征(DSSF/PSSF)。首先使用Rdkit将药物转换为半径为2的摩根指纹,再分别依据所有的药物、蛋白质结构信息分别生成两者的结构相似性矩阵,再分别在药物和蛋白质结构相似性矩阵上 获得 输入到Sim通道的单个药物和蛋白质的DSSF和PSSF。对于DSSF,它包含基准数据集中输入药物与所有药物的计算结构相似性评分。DSSF可以极大地捕获单个药物与基准数据集中所有药物的结构关联,从而能够更准确地表示输入的单个药物。获得每个药物和蛋白质的DSSF和PSSF之后就输入CNN进行提取降维特征(因为原始相似性特征的维数较大,部分相似性信息是多余的)。最后使用点积计算基于相似性的交互作用。
(1)生成药物结构相似性矩阵:根据谷本系数分别计算每个药物和数据集剩余每个药物的摩根指纹相似分数,这些值组成二维矩阵,即结构相似性矩阵。此时第i行值就表示第i个药物的结构相似特征向量。就建立起了一个药物与其他药物之间的联系。
(2)生成蛋白质相似性矩阵:利用距离【是一种用于度量两个字符串之间的相似性或差异的方法。它表示将一个字符串转换成另一个字符串所需的最小单字符编辑操作数。这些编辑操作可以是插入(在字符串中插入一个字符)、删除(删除一个字符)或替换(将一个字符替换为另一个字符)】来计算每个蛋白质的结构相似性。这些值组成二维矩阵,即结构相似性矩阵。此时第i行值就表示第i个蛋白质的结构相似特征向量。就建立起了一个蛋白质与其他蛋白质之间的联系。
2.子通道使用频繁连续子序列(FCS)方法,可适应提取蛋白质和药物的高质量大小的子结构。随后通过使用来学习子结构的信息和输入序列的化学语义信息(包括内容嵌入和位置嵌入),增强了子结构的上下文嵌入。在网络的最后一层使用CNN进行降维,以避免维数爆炸。最后使用点积计算基于子结构的交互作用。
3.交互预测模块(IP-)融合了上述两部分特征,通过展平以及串联特征来生成最终编码的交互特征,以便进行更准确的预测,通过全连接层获得预测DTI的概率得分。
1.药物表示
2.蛋白质表示
氨基酸序列
2.实验 1.数据集
(由于 仅包含正 DTI 对,因此按阳性样本的数量扩展负样本以平衡数据。在这项工作中,阴性样本被定义为药物 - 靶点对没有相互作用或未知其是否作用)和DAVIS。对于后续实验,按照数据集大小的100%、70%、50%和30%将数据集随机分为四个子数据集(为了测试在小数据集上的能力)。此外,选择70%的数据集用作训练集,10%作为验证集,20%用作测试集。
3.结果
消融实验:
1.验证在特征提取通道和 IP 模块之间使用 CNN 的必要性,需要对有和没有 CNN 的模型进行实验。观察到通过Sim通道和子通道提取的特征的维数非常大。因此,此时CNN的实现可以有效地去除冗余信息,获得具有更强相关性的特征,从而提高最终的预测性能。
2.验证子通道和Sim通道的必要性:只使用一个和交互模块来进行预测
单独使用子通道的预测性能优于单独使用Sim通道。但是,随着数据集大小的减小,预测性能会急剧下降,而 Sim 通道保持高稳定性。然而,当数据集大小从70%减小到30%时,子结构通道的ROC-AUC降低了7.3%,PR-AUC降低了8.8%。然而,数据集大小的减小对 Sim- 的影响很小,当数据集大小从 70% 减少到 30% 时,ROC-AUC 仅下降 3.2%,PR-AUC 下降 1.5%。这归因于Sim-可以使用其他分子的特征来确定自己的特征,从而在有限的数据上提取更多特征。
改进:使用药物和蛋白质的三维信息。
三.MCL-DTI: using drug and bi- cross- for drug–
利用药物多模态信息和双向交叉注意力学习方法预测药物-靶点相互作用 2023.8 三区
方法:用药物的分子图像和化学特征。药物的图像主要具有药物的结构信息和空间特征,而化学信息包括其功能和性质,可以相互补充,使药物表示更加有效和完整。同时,为了增强药物与靶点的交互特征学习,我们引入了双向多头注意力机制学习药物与靶点之间更深层次的语义关系,来提高DTI的性能。MSA 提取自注意力特征,MCA提取交互特征。
1.模型
整个模型主要由四个模块组成:特征编码器模块、特征解码器模块、特征融合模块和分类器。
1.特征编码器模块:
(1)药物:使用Rdkit工具包从序列中获取药物的图像和化学特征,用作药物的多模态表示。
对于图像特征的提取:CNN骨架模块提取局部特征包含卷积、批量归一化、激活和池化层;MSA 块包含层归一化 (LN)层、多头自注意层、MLP 块和残差连接,来提取语义特征。
对于文本信息的提取:文本信息通过用rdkit来操作得到的,是文本特征工厂,包括特征家族信息、特征类型信息和特征对应的原子信息;使用k−gram分割文本序列的方法从药物文本中提取信息,将嵌入表示馈送到MSA模块中,以获得化合物的文本特征。分配可学习的权重λ1和λ2给他们。权重越高,表明该模式对药物特征表示的影响较大。该药物被编码为Xdrug:
(2)蛋白质:使用其 FASTA 序列作为其文本信息。与药物的化学特征文本类似,首先通过k-gram获得其嵌入表示,然后获得目标的抽象特征,再通过 MSA 模块。
2.特征解码器模块:由两个解码器组成,每个解码器由一个MSA块和MCA块组成。MCA 块具有与 MSA 相同的 LN 层、MLP 块、残差连接。MSA和MCA的主要区别在于注意力输出的计算过程。MSA块旨在捕捉特征本身的内部关系,在计算注意力输出时,QKV都是通过线性矩阵变换从同一特征中获取的。MCA旨在捕获药物和靶标之间的相互作用信息。因此,对于药物解码器的MCA块,不仅需要输入药物特征,还需要输入蛋白质特征。对输入的药物特征进行矩阵线性变换,获取计算注意力输出所需的Q,对输入的蛋白质特征进行线性变换,获取KV。蛋白质解码器类似。使用药物解码器和蛋白质解码器,双向向对方发送各自的特征进行双向交叉学习,最后得到两个特征图。如下图的(a)
3.特征融合模块
接收来自两个解码器的特征图,按通道维度连接两个特征图,并将其馈送到融合块中。融合块包含一个二维卷积网络Conv2 D、一个 1D 卷积网络、一个 MLP 块和一个全连接层FC。我们通过卷积层提取级联的特征图,最后将它们馈送到FC层得到最终的预测结果P。如下图的(b)
1.药物表示
,再转化为文本信息和图信息
2.蛋白质表示
蛋白质FASTA序列(对蛋白质来说,它和氨基酸序列差不多)
2.实验 1.数据集
3.结果
1.实验结果
2.消融实验
共设置6种情况:
3.探讨了图像模态和文本模态的不同组合如何影响模型性能。使用多种方法组合进行实验。可以看到,当两个标量都设置并且都是可学习的时,性能是比较好的。
学习标量在实验的后期稳定,两个参数之间的比率是相对恒定的,标量的比率与ROC-AUC的值成反比,即当两个学习标量之间的差异较大时,模型的效率较低。
复现:
AUC PR-AUC
Human:
论文 0.987 0.989 0.961
复现 0.897 0.912 0.806
:
论文 0.992 0.994 0.959
复现 0.992 0.993 0.959
Davis:
论文 0.922 0.492 0.895
复现 0.922 0.480 0.881
四.: an graph-based model for drug- using NLP -level
一种基于可解释的基于图的模型,用于使用NLP句子级关系分类进行药物-靶点相互作用预测 2022.7 二区
该模型利用蛋白质结合位点和自我注意机制来解决药物 - 靶标相互作用预测的问题。通过识别对药物-靶标相互作用贡献最大的蛋白质结合位点来实现可解释性,是基于图的深度学习模型,它将小分子和蛋白质结合位点的结构特征以图的形式整合到DTI预测任务的管道中。类似于NLP中的句子分类问题,其中药物 - 靶标复合物被视为其生化实体(即蛋白质口袋和药物分子)之间具有关系意义的句子。
1.模型
模型由四个模块组成:数据准备、图嵌入学习模块、预测模块和解释模块。
1.在数据准备中,使用论文A for a ’s - 提出的算法找到蛋白质的结合位点(该算法计算蛋白质每个结合位点的边界框坐标。然后使用这些坐标将完整的蛋白质结构还原为肽片段的子集)。
2.下一步中,构建蛋白质口袋和配体的图并将其输入到拓扑自适应图CNN中,以从相应的图形中学习嵌入。
(1)蛋白质:提取蛋白质的结合位点之后,将它们表示为单独的图,其中每个原子都是一个节点,原子之间的连接是图中的边。每个原子的特征向量是1*31大小,包含原子类型、原子度、氢原子总数(连接)以及原子的隐式价、是否芳香等特征(都是用one-hot表示的)。由于同一个蛋白质可能具有不同的结合位点,因此对于同一蛋白质会生成多个不同的图,每个图代表蛋白质的不同结合位点。最终,这种方法会导致为同一蛋白质生成多个不同的嵌入,每个嵌入对应蛋白质的一个结合位点,从而能够捕捉不同位点的特征和性质。
(2)药物:根据构建一个双向图,构造包含原子类型,原子度,原子的形式电荷,原子的自由基电子数,原子的杂化,原子的芳香性和原子的总氢数的one-hot编码的特征向量,其维度为1*74。
(3)图注意力嵌入:使用拓扑自适应图CNN(TAGCN)(其输出是节点特征向量的串联),根据论文Gated graph 提出的方法对蛋白质和药物的图进行池化、提取嵌入操作(使用零填充将每个矩阵重塑为数据集中最大数量的结合口袋,并且蛋白质图的hops是4(代码里是2),药物图的hops是2)都是五次卷积。【TAGCN是图卷积网络的变体,卷积层的输出是由具有不同大小的滤波器得出的特征映射的加权和。TAGCN 的核心思想是通过特征传播来更新节点的表示。特征传播是通过邻接矩阵 A 来实现的。对于每个节点,TAGCN 会考虑其邻居节点(利用hops来决定考虑几步的邻居节点)的特征,并结合邻居节点的权重来更新节点的特征表示。并且引入了权重矩阵来学习节点之间的连接权重。这些权重矩阵是通过神经网络训练得到的,它们用于调整节点之间的信息传播。TAGCN 可以包含多个卷积层,每个卷积层都会重复上述的特征传播和权重矩阵学习步骤。这允许网络在多个层次上逐渐捕捉图数据的复杂关系。在卷积层之后,TAGCN 会得到每个节点的最终表示,这些表示包含了节点与其邻居的信息,并考虑了拓扑结构的自适应性。】
(4)然后将药物和蛋白质的特征载体进行整合:使用自注意力:在获得嵌入之后,将问题视为文本分类问题,并使用论文-based long short-term for 提出的方法来学习一个分类器:即双向LSTM+。使用双向LSTM,因为蛋白质结合位点和配体相互作用没有有意义的顺序。因此,序列中的未来上下文与过去上下文一样重要。最后使用逐元素相加来组合前进和后向传递的输出,其隐藏状态的大小设置为31。并且构建掩码的方式是每个结合位点只关注自身和相互作用的配体,以防止蛋白质的结合位点相互注意。最后的输出是被连成一维向量的特征。
3.预测模块:然后将串联的表示馈送到二元分类器中,用于预测DTI。使用具有Relu激活功能和两个全连接层的多层感知机MLP将提取的特征映射到最终分类输出中。此外,在每个线性层之前使用以获得更好的泛化性。
4.解释模块:网络中的自我注意机制使用药物-靶点对的串联嵌入作为输入来计算注意力输出,通过使模型学习蛋白质的哪些部分与给定药物-靶对中的配体相互作用来实现可解释性。
1.药物表示
根据构成的图
2.蛋白质表示
从序列中得到的蛋白质结合位点,被表示成图
2.实验
1.数据集
DUD-E数据集由来自八个蛋白质家族的102个靶标组成。每个靶标都有大约224种活性化合物和超过10000个decoy(诱饵,是化学性质相似而结构不相似的理论非活性化合物,这些诱饵的计算方式是其物理属性与活性化合物相似,但它们在拓扑上不同。即 是与蛋白质不发生相互作用的化合物,但是它和 与蛋白质发生作用的化合物的物理化学属性很相似。所以在实验中很难区分,可能会导致预测效果比较差)
使用了数据集的一小部分,该子集由39 747个阳性样本和31 218个阴性样本组成。此外,为了验证模型的泛化能力,将测试集分成两组蛋白质:在训练期间看到的蛋白质和模型未看到的蛋白质。
2.超参数
3.结果 1.实验结果:
数据集上:
2.消融实验:Bi-LSTM+Self-是论文的模型。比较了它和在图嵌入的最后一部分 分别只使用Bi-LSTM、Self-的模型
只使用Self-的效果在两个数据集上比较好。但是使用Bi-LSTM层在DUD-E数据集上实现了更高的精度,以捕获短程关系,然后使用Self-机制进一步提高特征的质量。由于其他两个数据集(Human和)不像DUD-E数据集那样具有挑战性,因此添加Bi-LSTM层不仅看起来没有优势,而且还可能导致模型过度拟合。